Projects
Analysis of hepatic zonation patterns from whole-slide images
|
|

This project explores the critical interplay between lobular geometry and the zonated distribution of cytochrome P450 (CYP) enzymes across species. We present an innovative approach to assess lobular geometry and zonation patterns using whole slide imaging (WSI). This method allows a detailed, systematic comparison of lobular structures and spatial distribution of key CYP450 enzymes and glutamine synthetase in four different species (mouse, rat, pig, and human). Our results shed light on species differences in lobular geometry and enzymatic zonation, providing critical insights for drug metabolism research. Based on our approach we could determine the minimum number of lobules required for a statistically representative analysis, an important piece of information when evaluating liver biopsies and deriving information from WSI.
Physiologically based pharmacokinetic (PBPK) model of creatinine and inulin
|
|
 model of creatinine and inulin")
The glomerular filtration rate (GFR) is a measure of how well your kidneys are filtering waste and excess fluids from your blood. It is an important indicator of kidney function and is expressed in milliliters per minute (mL/min). GFR is calculated based on factors such as age, gender, body size, and creatinine levels in the blood. It is important to monitor GFR in individuals with kidney disease or at risk of developing kidney disease to ensure appropriate treatment and management of the condition.
In this project, we aim to create a physiologically based pharmacokinetic (PBPK) model for inulin and creatinine to assess the glomerular filtration rate (GFR). Our goal is to gain a deeper understanding of the impact of various inulin protocols on GFR measurement and the influence of physiological factors, such as body composition, on GFR determination using inulin. Furthermore, it should enhance our understanding of how variations in creatinine production, muscle mass, and tubular secretion of creatinine influence eGFR calculations.
Physiologically based pharmacokinetic (PBPK) model of CYP2D6 polymorphism
|
|
|
Grzegorzewski, J., Brandhorst, J., König, M. Physiologically based pharmacokinetic (PBPK) modeling of the role of CYP2D6 polymorphism for metabolic phenotyping with dextromethorphan Front Pharmacol. 2022 Oct 24;13:1029073 |
 model of CYP2D6 polymorphism")
Physiologically based pharmacokinetic (PBPK) model of dextromethorphan
The cytochrome P450 2D6 (CYP2D6) is a key xenobiotic-metabolizing enzyme involved in the clearance of many drugs. Genetic polymorphisms in CYP2D6 contribute to the large inter-individual variability in drug metabolism and could affect metabolic phenotyping of CYP2D6 probe substances such as dextromethorphan (DXM).
To study this question, we (i) established an extensive pharmacokinetics dataset for DXM; and (ii) developed and validated a physiologically based pharmacokinetic (PBPK) model of DXM and its metabolites dextrorphan (DXO) and dextrorphan O-glucuronide (DXO-Glu) based on the data. Drug-gene interactions (DGI) were introduced by accounting for changes in CYP2D6 enzyme kinetics depending on activity score (AS), which in combination with AS for individual polymorphisms allowed us to model CYP2D6 gene variants. Variability in CYP3A4 and CYP2D6 activity was modeled based on in vitro data from human liver microsomes.
Both, model and database are freely available for reuse.Protein distributions in the human liver
|
|

Individual variations in the clearance of test substances via specific enzymes and transporters in the liver are substantial. These variations are attributed to genetic polymorphisms and protein abundance in the liver. Understanding protein distributions is an essential input for computational models, such as physiologically based pharmacokinetic (PBPK) models, and for metabolic phenotyping, such as liver function tests, to study drug detoxification.
The primary objective of this project is to provide a comprehensive overview of the variability in protein abundance among CYP and UGT enzymes as well as ABC and SLC transporter isoforms in human livers. Furthermore, this project aims to examine relationships between enzymes and transporters, such as correlations, to gain insight into the factors affecting individual drug metabolism. This systematic analysis of proteins will provide a valuable resource for computational modeling of drug detoxification by the liver and enable a better understanding of individualized drug responses.
ATLAS - AI and Simulation for Tumor Liver ASsessment
|
|

Liver cancer is the second most common cause of cancer-related death. Diagnosis and treatment are time-critical and require highly patient-specific diagnostic and treatment pathways. Medical decision-making is based on a variety of interdependent factors related to different medical disciplines, past experience and clinical guidelines. Taking into account all decision factors in combination with the possible therapeutic approaches is a major challenge for physicians and often cannot be solved optimally even in an interdisciplinary tumor board.
In this project, we are developing ATLAS, a decision support tool that will significantly assist clinicians in meeting this challenge. Based on AI methods, ATLAS processes all relevant patient data from databases, systems medicine and continuum biomechanical in silico prognosis models as well as individual patient data. The tool is being developed in a co-design approach by experts in surgical oncology, mathematical modeling and machine learning. The selected technologies will integrate automated understanding of a highly complex patient situation through simulation of liver functions with expert knowledge and ontology-driven learning with knowledge graphs from retrospective liver tumor cases.
ATLAS will be based on a detailed historical data cohort of more than 6,000 patients with liver tumors and will be evaluated on case studies at the University Hospital of Jena. The integration of medical expert knowledge, mathematical modeling and artificial intelligence represents a highly original and promising approach for high-quality diagnosis and treatment of liver tumors, resulting in patient-specific improvement of prognosis. The scientific knowledge gained from these projects will provide opportunities for transfer to malignancies in other organs, such as the lung, kidney or brain. The development of tools and demonstrators will provide sustainable exploitation pathways for future commercial applications.
Hepatic zonation patterns of Cytochrome P450 enzymes
|
|

Quantification of hepatic zonation patterns from Immunofluorescence and Immunohistochemistry Images
This project implements an image analysis pipeline based on whole slide images (WSI) from brightfield or fluorescence microscopy. The zonation patterns will be quantified.
The developed pipeline will be applied to study species differences in zonation patterns of CYP enzymes and changes in zonation due to hepatic steatosis and perfusion changes.
Simvastatin therapy in different subtypes of hypercholesterolemia
|
|
|
F. Bartsch, J. Grzegorzewski, H. Pujol, HM. Tautenhahn, M. König Simvastatin therapy in different subtypes of hypercholesterolemia - a physiologically based modelling approach medRxiv 2023.02.01.23285358 |

Simvastatin therapy in different subtypes of hypercholesterolemia
Hypercholesterolemia is a multifaceted plasma lipid disorder with heterogeneous causes including lifestyle and genetic factors. A key feature of hypercholesterolemia is elevated plasma levels of low-density lipoprotein cholesterol (LDL-C). Several genetic variants have been reported to be associated with hypercholesterolemia, known as familial hypercholesterolemia (FH). Important variants affect the LDL receptor (LDLR), which mediates the uptake of LDL-C from the plasma, apoliporotein B (APOB), which is involved in the binding of LDL-C to the LDLR, and proprotein convertase subtilisin/kexin type 9 (PCSK9), which modulates the degradation of the LDLR. A typical treatment for hypercholesterolemia is statin medication, with simvastatin being one of the most commonly prescribed statins.
In this project, the LDL-C lowering therapy with simvastatin in hypercholesterolemia was investigated using a computational modeling approach. A physiologically based pharmacokinetic model of simvastatin integrated with a pharmacodynamic model of plasma LDL-C (PBPK/PD) was developed based on extensive data curation. A key component of the model is LDL-C turnover by the liver, consisting of: hepatic cholesterol synthesis with the key enzymes HMG-CoA reductase and HMG-CoA synthase; cholesterol export from the liver as VLDL-C; de novo synthesis of LDLR; transport of LDLR to the membrane; binding of LDL-C by LDLR via APOB; endocytosis of the LDLR-LDL-C complex; recycling of LDLR from the complex. The model was applied to study the effects of simvastatin therapy in hypercholesterolemia due to different causes in the LDLR pathway corresponding to different subtypes of hypercholesterolemia.
Model predictions of LDL-C lowering therapy were validated with independent clinical data sets. Key findings are: (i) hepatic LDLR turnover is highly heterogeneous among FH classes; (ii) despite this heterogeneity, simvastatin therapy results in a consistent reduction in plasma LDL-C regardless of class; and (iii) simvastatin therapy shows a dose-dependent reduction in LDL-C. Our model suggests that the underlying cause of hypercholesterolemia does not influence simvastatin therapy. Furthermore, our model supports the treatment strategy of stepwise dose adjustment to achieve target LDL-C levels. Both the model and the database are freely available for reuse.Computational Modeling of Dynamical Liver Function Tests: LiMAx
|
|
|

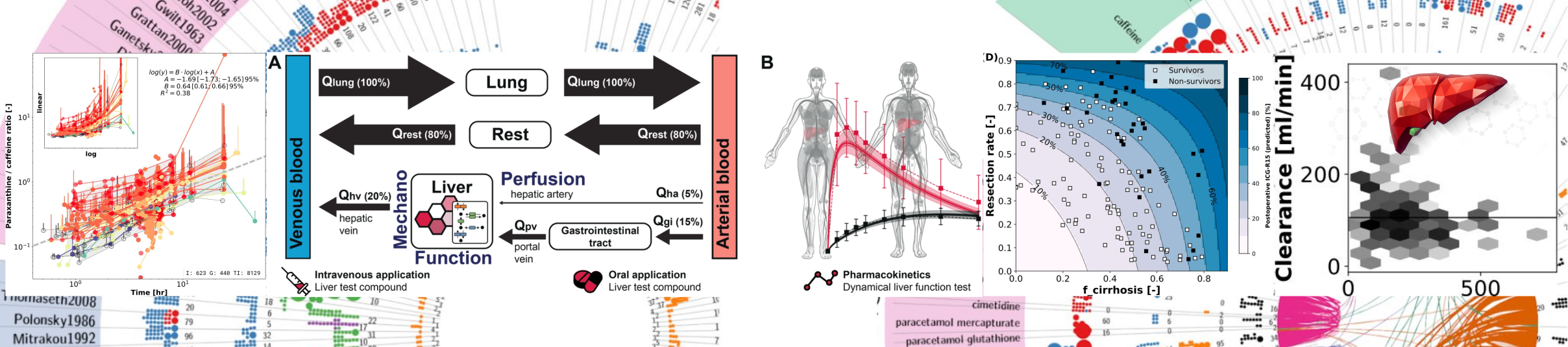
Modeling LiMAx liver function test
Assessment of liver function is a key task in hepatology but accurate quantification of hepatic function has remained a clinical challenge. Dynamic liver function tests are a promising tool for the non-invasive evaluation of liver function in vivo. One class of such tests are breath tests based on the conversion of 13C-labeled substrates by the liver to 13CO2 subsequently measured in the breath. A commonly applied substrate is 13C-methacetin, converted to paracetamol and 13CO2 via cytochrome P450 1A2 (CYP1A2), used orally in the methacetin breath test (MBT) and intravenously in the LiMAx test. An important clinical question is which factors can affect MBT and LiMAx results. The aim of our study was to answer this question using computational modeling to derive basic information for a better understanding of the methacetin breath test and factors influencing its results.
A physiological based pharmacokinetics (PBPK) model for 13C-methacetin breath tests including absorption, distribution, metabolism and elimination of 13C-methacetin, paracetamol, and 13C-bicarbonate/13CO2 was developed. The model correctly predicts data from more than 20 clinical studies after oral and intravenous application under various dosing regimens of paracetamol, 13C-bicarbonate, and 13C-methacetin, based on retrospective analysis. Model predictions were validated based on data from multiple studies consisting of 13C-bicarbonate kinetics, LiMAx dosing study and retrospective analysis of LiMAx data from healthy smokers and non-smokers. Sensitivity analysis was performed to identify key factors influencing MBT and LiMAx.
The model was applied to study the effect of clinically relevant parameters like CYP1A2 content, hepatic perfusion or lifestyle factors like smoking on LiMAx. In summary, we present a valuable tool for the evaluation of dynamical liver function tests based on 13C-methacetin.Reproducible simulation studies targeting COVID-19
|
|
|
F. Schreiber, B. Sommer, G. Bader, P. Gleeson, M. Golebiewski, T. Gorochowski, M. Hucka, S. Keating, M. König, C. Myers, D. Nickerson, D. Walthemath Specifications of Standards in Systems and Synthetic Biology:Status and Developments in 2020 J Integr Bioinform. 2020 Jun 29;17(2-3):20200022 |

Interoperable COVID-19 models
In a collaboration between the University of Greifswald, the Humboldt-University Berlin, code ahoi, and the BioModels database at EMBL-EBI, we aim to rapidly disseminate simulation studies of COVID-19 models to the research community, in interoperable formats and in high quality.
This project is supported by EOSCsecreteriat.eu COVID-19 Fast Track Funding.
Modeling Hepatic Central Metabolism
|
|
|
Berndt N., Bulik S., Wallach I., Wünsch T., König M., Stockmann M., Meierhofer M., Holzhütter HG. HEPATOKIN1: A Biochemistry-Based Model of Liver Metabolism Suited for Applications in Medicine and Pharmacology Nat Commun. 2018 Jun 19;9(1):2386. |

Modeling hepatic metabolism in health and disease
The epidemic increase of non-alcoholic fatty liver diseases (NAFLD) requires a deeper understanding of the regulatory circuits controlling the response of liver metabolism to nutritional challenges, medical drugs, and genetic enzyme variants. As in vivo studies of human liver metabolism are encumbered with serious ethical and technical issues, we were involved in the development of a comprehensive biochemistry-based kinetic model of the central liver metabolism including the regulation of enzyme activities by their reactants, allosteric effectors, and hormone-dependent phosphorylation.
Stratification and personalization of computational models
|
|
|
|

Workflow for extraction of pharmacokinetics data
Physiologically based pharmacokinetics (PBPK) models are an important tool for understanding dynamical liver function tests. Such models allow to simulate the absorption, distribution, metabolization and elimination (ADME) of test substances. Stratification and personalization of PBPK models is an important task in systems biology and systems medicine, e.g., to simulate different subpopulations like smokers vs. non-smokers, obese vs. lean people, or children vs. adults vs. elderly, or to simulate the individual person based for instance on anthropometric information like age, gender or bodyweight. Such PBPK models can be described in standard exchange formats like the systems biology markup language (SBML) which allow a reproducible, exchangable and interoperable encoding of these models. On the other hand data sets for stratification and personalization of such models are available from the research literature or clinical cooperation partners. An open task to implement an workflow for the stratification and personalization of PBPK models from existing datasets. This includes the automatic extraction of information via literature mining and machine learning, storage of information in a pharmacokinetics database, and parametrization of models base on subsets of information.
Multi-scale Model of Hepatic Metabolism
|
|
|

Multiscale model of human galactose metabolism - from single hepatocytes to individual liver function
Understanding how liver function arises from the complex interaction of morphology, perfusion, and metabolism from single cells up to the entire organ requires systems-levels computational approaches. We report a multiscale mathematical model of the Human liver comprising the scales from single hepatocytes, over representation of ultra-structure and micro-circulation in the hepatic tissue, up to the entire organ integrated with perfusion. The model was validated against data on multiple spatial and temporal scales. Herein we describe the model construction and application to hepatic galactose metabolism demonstrating its utility via i) the personalization of liver function tests based on galactose elimination capacity (GEC), ii) the explanation of changes in liver function with aging, and iii) the prediction of population variability in liver function based on variability in liver volume and perfusion. We conclude that physiology- and morphology-based multiscale models can improve the evaluation of individual liver function.
Liver Function Tests
|
|
|

Personalized evaluation of liver function tests
Understanding how liver function results from the complex interaction of hepatic morphology, perfusion and metabolism across multiple spatial scales, from the cellular level up to the entire organ, requires systems levels computational approaches. Despite the importance of quantifying liver function in the clinics, currently no computational model of the human liver is able to utilize the morphological and physiological information from imaging methods for a personalized evaluation of quantitative function tests. Our objectives are
- providing an open, freely available and reproducible multiscale model of the human liver which can readily be employed to evaluate the hepatic clearance of various test substances used in quantitative liver function tests in the clinics
- providing the infrastructure and methods to integrate experimental and clinical data on all scales (imaging data, anthropomorphic information, perfusion data multipleindicator dilution data, microcirculation and ultrastructure parameters, protein amounts, ...)
- create metabolic modules for the elimination of various test substances and drugs (methacetin, galactose, BSP, ICG, midazolam, codeine, caffeine, torsemide, pravastatin, talinolol) by the liver. This will allow the analysis of a wide range of clinical studies and create an integrative platform for the clearance of these substances by the liver.
Biomarker signatures in liver disease
|
|
|
Kerstin A.Δ, König M.Δ, Hoppe A., Thomas M., Müller I., Ebert M., Weng H., Holzhütter HG., Zanger UM., Bode J., Vollmar B. and Dooley S. (Δ equal contribution) Pathobiochemical signatures of cholestatic liver disease in bile duct ligated mice BMC Syst Biol. 2015 Nov 20;9(1):83. |

Pathobiochemical signatures of cholestatic liver disease
Disrupted bile secretion leads to liver damage characterized by inflammation, fibrosis, eventually cirrhosis, and hepatocellular cancer. As obstructive cholestasis often progresses insidiously, markers for the diagnosis and staging of the disease are urgently needed. To this end, we compiled a comprehensive data set of serum markers, histological parameters and transcript profiles at 8 time points of disease progression after bile duct ligation (BDL) in mice, aiming at identifying a set of parameters that could be used as robust biomarkers for transition of different disease progression phases.
High Performance Simulators and Simulation Software
|
|
|
Medley K., Choi K., König M., Smith L., Gu S., Hellerstein J., Sealfon S., and Sauro HM. Tellurium Notebooks — An Environment for Reproducible Dynamical Modeling in Systems Biology. PLoS Comput Biol. 2018 Jun 15;14(6):e1006220 |

Tellurium Notebooks - An Environment for Dynamical Model Development, Reproducibility, and Reuse
The considerable difficulty encountered in reproducing the results of published dynamical models limits validation, exploration and reuse of this increasingly large biomedical research resource. To address this problem, we have developed Tellurium Notebook, a software system that facilitates building reproducible dynamical models and reusing models by 1) supporting the COMBINE archive format during model development for capturing model information in an exchangeable format and 2) enabling users to easily simulate and edit public COMBINE-compliant models from public repositories to facilitate studying model dynamics, variants and test cases. Tellurium Notebook, a Python--based Jupyter--like environment, is designed to seamlessly inter-operate with these community standards by automating conversion between COMBINE standards formulations and corresponding in--line, human--readable representations. Thus, Tellurium brings to systems biology the strategy used by other literate notebook systems such as Mathematica. These capabilities allow users to edit every aspect of the standards--compliant models and simulations, run the simulations in--line, and re--export to standard formats. We provide several use cases illustrating the advantages of our approach and how it allows development and reuse of models without requiring technical knowledge of standards. Adoption of Tellurium should accelerate model development, reproducibility and reuse.
Reproducible simulation experiments
|
|
|

tellurium-web: An online database for reproducible simulation experiments in computational biology
We present tellurium-web, a free and open online database and simulation tool for reproducible simulation experiments in computational biology. Tellurium-web is build on top of existing standards in computational biology, i.e., the SBML for models, SED-ML for description of simulation experiments and the COMBINE archive format for exchange of simulation studies. Key features are 1) large set of example archives (e.g., simulation studies from biomodels, SED-ML specification examples, SBML testsuite, JWS online, tellurium examples); 2) COMBINE archive browser with access to metadata and content of the archives; 3) Execution of archives using a high-performance simulator (libroadrunner) integrated into modeling platform for computational biology (tellurium); 4) REST web services for getting, posting and modifying archives providing means for simple integration into computational workflows; and 4) filtering of archives based on tags and users.
FluType: Influenza Classification based on Machine Learning
|
|
|

Development of an influenza sub-classification platform based on peptides
The FluTypeDB project is a web application for the data management of binding assays for the classification of influenza subtypes. FluTypeDB consists of a database and web interface with focus on various binding assays for the classification of influenza viruses and contains experimental data based on
- ELISA antibody binding assays
- Peptid binding assays on microwell plates
- Peptid bindings assays on peptide microarrays
Visualization of kinetic data for modeling
|
|
|
König M. cy3sabiork: A Cytoscape app for visualizing kinetic data from SABIO-RK [version 1; referees: awaiting peer review]. F1000Research 2016, 5:1736 |

Cytoscape app for visualizing kinetic data from SABIO-RK
Kinetic data of biochemical reactions are essential for the creation of kinetic models of biochemical networks. One of the main resources of such information is SABIO-RK, a curated database for kinetic data of biochemical reactions and their related information. Despite the importance for computational modelling there has been no simple solution to visualize the kinetic data from SABIO-RK. In this work, I present cy3sabiork, an app for querying and visualization of kinetic data from SABIO-RK in Cytoscape. The kinetic information is accessible via a combination of graph structure and annotations of nodes, with provided information consisting of: (I) reaction details, enzyme and organism; (II) kinetic law, formula, parameters; (III) experimental conditions; (IV) publication; (V) additional annotations. cy3sabiork creates an intuitive visualization of kinetic entries in form of a species-reaction-kinetics graph, which reflects the reaction-centered approach of SABIO-RK. Kinetic entries can be imported in SBML format from either the SABIO-RK web interface or via web service queries. The app allows for easy comparison of kinetic data, visual inspection of the elements involved in the kinetic record and simple access to the annotation information of the kinetic record. The cy3sabiork approach was applied in the computational modelling of galactose metabolism in the human liver.